Alexandra Andrade1, Carolina Ferreira Gonçalves2, Pedro Vale Fernandes2, Teresa Jacinto3, Filomena Teixeira3, Edite Costa3, Paulo Rego Sousa4.
1Department of Pediatrics, Hospital Central do Funchal, Funchal, Portugal,
2Department of Dermatology, Hospital Central do Funchal, Funchal, Portugal,
3Department of Neonatal and Pediatric Intensive Care, Hospital Central do Funchal, Funchal, Portugal,
4Pediatric Neurology Unit, Department of Pediatrics, Hospital Central do Funchal, Funchal, Portugal.
ADDRESS FOR CORRESPONDENCE
Alexandra Andrade, PServiço de Pediatria, Av. Luís de Camões 6180, 9000-177 Funchal.
Email: asgalg@gmail.com | | Abstract | | Incontinentia pigmenti is a rare X-linked genodermatosis caused by mutations in the IKBKG gene. It is a systemic disease mainly involving tissues of ectodermic origin, manifesting itself primarily as skin lesions but also as changes in the hair, nails, teeth, breasts, eyes and nervous system. We present a case of a newborn who presented with a vesicular rash in the first week of life. Histology revealed typical eosinophilic infiltrate and spongiosis in the biopsy sample. The mother had hypopigmented lesions compatible with the same diagnosis, later confirmed in both cases by genetic testing. Although rare, incontinentia pigmenti should be considered in a child presenting vesicular and verrucous cutaneous lesions. Timely diagnosis is a must for establishing adequate follow-up. | | | | Keywords | | Cutaneous manifestations, Incontinentia pigmenti, NEMO/IKBKG, Genodermatosis, Systemic disease | | | | Introduction | Incontinentia pigmenti (IP) or Bloch-Sulzberger syndrome, is a rare X-linked genodermatosis, with an estimated prevalence at birth of 1.2:100 000.1 It is associated with the IKBKG gene (formerly known as NEMO) and is usually lethal in males. Its phenotypical expression is variable, ranging from mild to severe. Virtually all patients present with skin lesions, but other symptoms are less common and more diverse, ranging from changes in the skin of the appendages to dental, ocular and neurological symptoms.2
In this report, we describe a case of IP in a newborn with no known family history, presenting with cutaneous lesions, that later led to the same diagnosis in her mother.
| | | | Case Report | A five-day-old female newborn was observed in our emergency department due to a generalized vesicular rash. She had no other complaints.
The baby was born at 39 weeks after an uneventful pregnancy to an apparently healthy 34-year-old mother. During clinical evaluation, the child appeared to be well. She presented a vesicular dermatosis overlaying an erythematous base on the trunk and extremities, with linear distribution along the Blaschko lines (see Figure 1). According to the mother, a few smaller cutaneous lesions were present since birth but grew more noticeable by the fourth and fifth day of age. No other signs were noted. The remaining physical examination was normal.
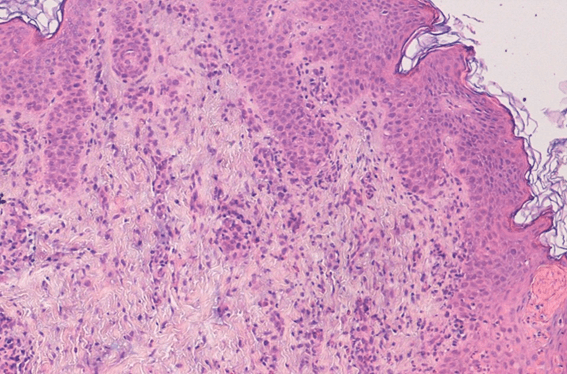
Due to the typical presentation and linear distribution following the Blaschko lines, a clinical diagnosis of IP was suggested. A confirmatory skin biopsy revealed typical reactive changes in the epidermis showcasing a polymorphic infiltrate rich in eosinophils with intra-epidermal microvesiculation and spongiosis as well as an abundant inflammatory dermal infiltrate, also rich in eosinophils, both in interstitial and perivascular locations (Figure 2).
Figure 1. Vesicular lesions over an erythematous base with linear distribution along the Blaschko lines.
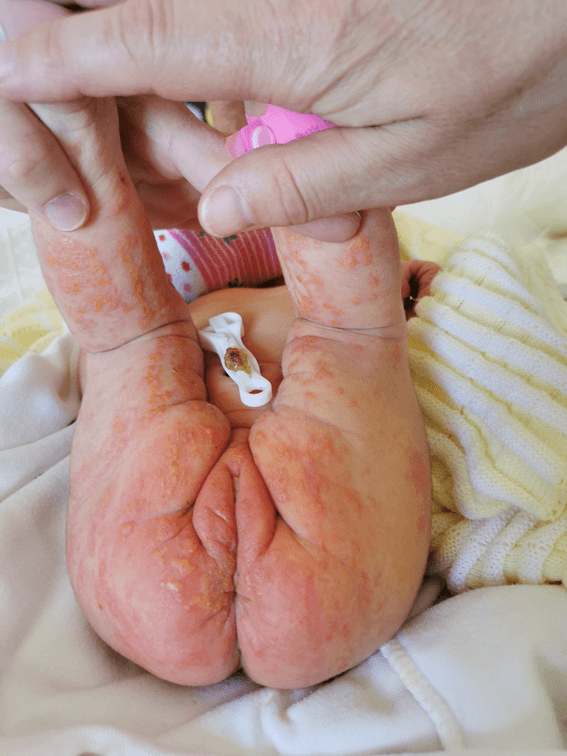
Figure 2. Polymorphic infiltrate rich in eosinophils, with intra-epidermal microvesiculation and spongiosis as well as an abundant inflammatory dermal infiltrate.
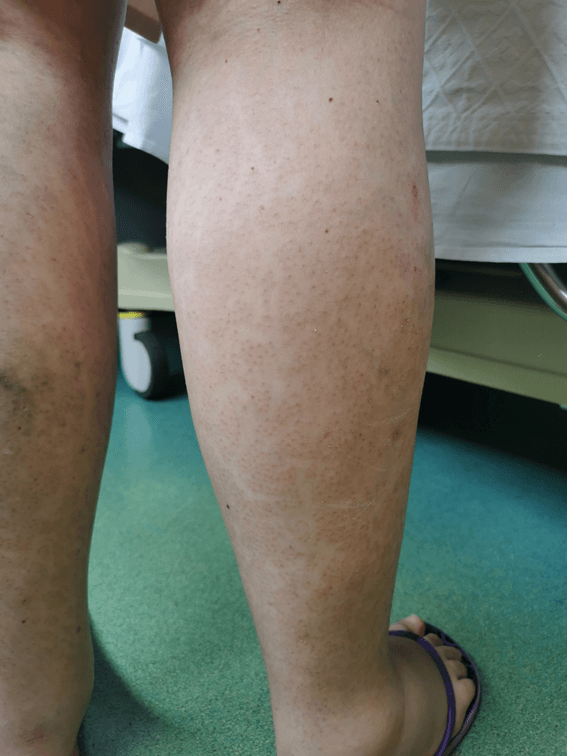
On further clinical investigation, the mother herself presented hypopigmented linear maculae on her legs and alopecia (Figure 3). Furthermore, there was an underestimated history of hypodontia, myopia, astigmatism and divergent strabismus earlier on, as well as past miscarriage (twice, at seven and sixteen weeks of gestation). A fetal autopsy of the latest miscarriage at 16 weeks showed a male fetus with anomalies in the face and limbs, pulmonary hypoplasia and fetal growth restriction. Although no further diagnostic studies were carried out post mortem, these aspects are compatible with IP. The remaining family history was irrelevant. Genetic studies confirmed the presence of deletions of exons 4-10 in the IKBKG gene in both the mother and child, confirming both the clinical diagnosis and the transmission of the mutation within the family.
Figure 3. Hypopigmented linear maculae on the mother’s legs with alopecia.
During follow-up, the skin lesions evolved to the verrucous stage, then to the hyperpigmented phase and eventually faded, which is the usual evolution in these cases. Psychomotor development has been normal so far. The child maintains in multidisciplinary follow up in Dermatology, Pediatric Neurology, Ophthalmology and General Pediatrics. | | | | Discussion | IP is of dominant X-linked inheritance and its most frequent mutation is the deletion of exons 4 through 10 of the IKBKG gene, localized on chromosome Xq28, in about 70%-80% of patients. The IKBKG gene product is responsible for the activation of nuclear factor kappa B (NF-kB), which is a regulatory protein involved in cellular apoptosis, inflammation and immunity. Its phenotypic expression is highly variable, even among blood-related patients. This is thought to be associated with skewed X-inactivation, which predominantly eliminates cells bearing the mutated IKBKG gene.3
The majority of patients present with skin lesions. These are classically described as evolving in four stages, following a specific pattern, known as the lines of Blaschko. The vesicular stage is characterized by marked erythema with linear vesicles and pustules and is often present at birth. The verrucous stage presents with wart-like verrucous papules, generally within the first weeks or months of life. The hyperpigmented stage, in which the patient has linear or swirling patches of hyperpigmentation, presents in infancy and adolescence. The hypopigmented stage consists usually of streaks or whirls of hypopigmentation, often associated with cutaneous atrophy and alopecia and usually occurs in adulthood, like the ones present in the mother of our case. Not all patients experience the four stages and some can present more than one at the same time.4 Our patient experienced mostly stage 1 and 2 lesions that for a time appeared simultaneously, which is the most typical presentation. At that stage, other diagnoses that can be considered are mainly infectious, such as infection by herpes simplex virus, varicella-zoster virus and staphylococcus aureus.
The skin of the appendages can also change, manifesting in a broad spectrum of symptoms such as alopecia, coarse or brittle hair, nail dystrophy and nailbed keratotic tumors in 28%-40% of patients. Anomalies of the mammary tissue are less common, occurring in around 11% of patients. Hypodontia, microdontia, delayed eruption or abnormally shaped teeth are also ectodermal symptoms of IP and can occur in up to 54% of patients.5 Central nervous system (CNS) anomalies were found in 13%-35% of patients, consisting primarily of seizures, neurocognitive impairment, stroke and microcephaly6 and 37% of patients suffered from ocular anomalies, most frequently retinal anomalies.7 Other rare symptoms have been described in association with IP, like skeletal and cardiopulmonary anomalies.
In the present case, not only were we able to identify the infant’s presentation but also the clinical manifestations in the adult mother consisting of not only atrophic chronic skin changes but also a plethora of systemic indications, supporting our diagnosis at a different ages and stages. Skin biopsy can be helpful when obtained during stage 1, in which the skin has characteristic histopathologic findings such as eosinophilic spongiosis, intra-epidermal vesicles containing eosinophils and apoptotic keratinocytes in the epidermis.8
Diagnosis criteria consist of the several stages of skin lesions as the major criteria and, for the minor criteria, anomalies such as dental, ocular, CNS, breast, abnormal hair or nails, multiple male miscarriages and IP pathohistological findings. A definitive diagnosis is established when genetic testing demonstrates the deletion of exons 4-10 or another known pathogenic mutation in IKBKG associated with IP.
This case presented the most common mutation occurring in IP, but we aim to emphasize the importance of the family history, which allowed us to make a belated diagnosis in a blood-related female adult. Thus, referral of the mother to genetic counselling and pre-implantation genetic testing in further gestations was recommended henceforth.
Management of IP is symptomatic since etiological therapeutic targeting is not presently available. According to the degree of severity of the phenotype of the patient, management may involve a multidisciplinary care team. Vesicular lesions require gentle care with emollients and monitoring for secondary skin infection. Ophthalmologic surveillance is recommended in all confirmed or suspected IP cases, for eye complications can present silently without proper monitoring. Follow-up with a dental consultation should be required. If significant CNS involvement is present, close evaluation by a pediatric neurologist specialist is necessary. Genetic counseling should be offered to the patient and their family.
In conclusion, IP is a multisystemic disorder and a challenging diagnosis, both due to its rarity and the variability of symptoms that can be present. An adequate and timely diagnosis is crucial to establish the correct follow-up of the patient. Follow-up of ophthalmologic sequels and neurodevelopmental impairment are some of the most important features to target in patients with an early diagnosis and/or those who only present skin lesions. Family history and clinical evaluation of suggestive complaints or signs in first-degree relatives can also yield useful information pertaining to future management. | | | | Compliance with Ethical Standards | | Funding None | | | | Conflict of Interest None | | |
- Prevalence and incidence of rare diseases: Bibliographic data [Internet]. Orphanet Report Series. 2022 [cited 2022 Apr 25]. Available from: http://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_alphabetical_list.pdf
- Minić S, Trpinac D, Obradović M. Incontinentia pigmenti diagnostic criteria update. Clin Genet. 2014;85(6):536-42. [CrossRef]
- Fusco F, Bardaro T, Fimiani G, Mercadante V, Miano MG, Falco G, et al. Molecular analysis of the genetic defect in a large cohort of IP patients and identification of novel NEMO mutations interfering with NF-κB activation. Hum Mol Genet. 2004 Aug 15;13(16):1763-73. [CrossRef]
- Swinney CC, Han DP, Karth PA. Incontinentia pigmenti: A comprehensive review and update. Vol. 46, Ophthalmic Surgery Lasers and Imaging Retina. Slack Incorporated; 2015. p. 650-7. [CrossRef]
- Greene-Roethke C. Incontinentia Pigmenti: A Summary Review of This Rare Ectodermal Dysplasia With Neurologic Manifestations, Including Treatment Protocols. Journal of Pediatric Health Care. 2017 Nov 1;31(6):e45-52. [CrossRef]
- Meuwissen MEC, Mancini GMS. Neurological findings in incontinentia pigmenti; a review. Vol. 55, European Journal of Medical Genetics. 2012. p. 323-31. [CrossRef]
- Minić S, Obradović M, Kovačević I, Trpinac D. Ocular anomalies in incontinentia pigmenti: Literature review and meta-analysis. Srp Arh Celok Lek. 2010;138(7-8):408-13. [CrossRef]
- Poziomczyk CS, Maria FDS, Freitas AM, Fiegenbaum M, Bau AEK, Recuero JK, et al. Incontinentia pigmenti. An Bras Dermatol. 2014;89(1):26-36. [CrossRef]
DOI: https://doi.org/10.7199/ped.oncall.2025.70
|
| Cite this article as: | | Andrade A, Gonçalves C F, Fernandes P V, Jacinto T, Teixeira F, Costa E, Sousa P R. Incontinentia Pigmenti - one case, two diagnoses. Pediatr Oncall J. 2025;22. doi: 10.7199/ped.oncall.2025.70 |
|