How Complement Fights Infections?
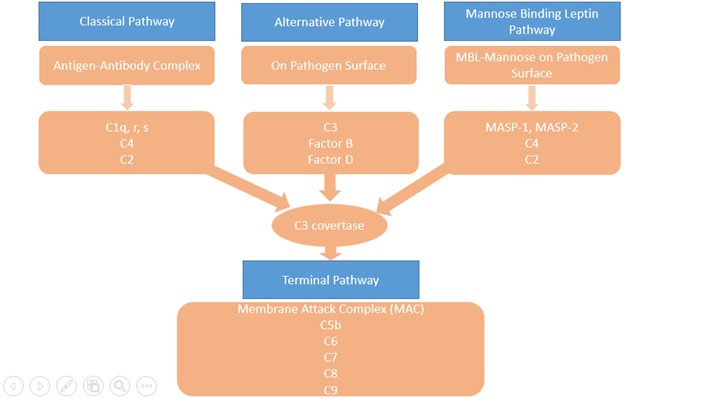
The complement system consists of more than 30 proteins, present in blood and tissues, as well as other proteins anchored on the surfaces of cells. The primary functions of the complement system are to protect from infection, to remove particulate substances, (like damaged or dying cells, microbes, or immune complexes), and to help modulate adaptive immune responses. As part of the innate immune system, complement acts immediately to start the process of removal and resolution of the problem. The complement works with the inflammatory cells of the innate immune system and those of adaptive or acquired immunity. Complement proteins in the circulation are not activated until triggered by an encounter with a bacterial cell, a virus, an immune complex, damaged tissue, or other substance not usually present in the body. There are 3 major activations of complement pathways; classical (CP), alternative(AP), and leptin pathway(LP) summarized in Figure 1. The Classical Pathway (CP) is activated primarily by immunoglobulins that are bound to antigens. Aggregates of immunoglobulins such as cryoglobulins also activate the CP. Components of the CP are C1q, C1r, C1s, C2, and C4. The Lectin Pathway (LP) is initiated by Mannose-binding lectin (MBL) or one of the Ficolins to a target molecule. Both MBL and ficolins are present in the complex with MBL-associated serine protease(MASP) and activate C4 and C2. The Alternative Pathway (AP) is initiated by fragments of the complement component C3. Factor B, Factor D, and properdin also involved in AP. Properdin, the only positive regulator in the complement system available in AP. Properdin makes it possible for the amplification loop of the alternative pathway to set up a very efficient mechanism for putting lots of C3b onto the surface of the activating cells, protein complexes, or particles in the immediate vicinity of the activation site. These 3 pathways converge at the component C3. Although each branch is triggered differently, the common goal is to deposit clusters of C3b on a target. This deposition provides for assembly of the membrane attack complex (MAC).
The Terminal Pathway (TP) is the final set of steps in the complement activation process that forms a membrane lesion or hole (membrane attack complex or MAC) that kills susceptible bacteria or other cells that activate complement on their surfaces. The TP is dependent upon at least one of the other pathways to initiate the process that it then completes. The components of the TP are C3, C5, C6, C7, C8, and C9. The form of the MAC, called the Terminal Complement Complex (TCC) can be found in the circulation after complement activation occurs and makes a useful laboratory marker for complement activation.
Figure 1: Complement Pathways
Introduction
The complement system is part of the immune system, whose major function is to protect the host from infections. In the defense, both the innate and the adaptive immune systems are operative through several mechanisms. The complement system comprises an important part of the innate defense and some act bridging innate and adaptive immunity (Dunkelberger et al 2010). It promotes the inflammatory response, eliminates pathogens, and enhances the immune response. Deficiencies in the complement cascade can lead to overwhelming infection and sepsis (JE Figueroa, Denssen 1991). In addition to playing an important role in host defense against infection, the complement system is a mediator in both the pathogenesis and prevention of immune complex disease, such as systemic lupus erythematosus.
The importance of complement in defense against bacterial infections has become clear with some role in against fungal and parasitic infection (Hirsch RL et al 1981). The ability of Mannose-binding lectin bind to HIV has been shown in the in-vitro study (Saifuddin et al 2000).
Complement deficiencies are rare. However, they are poorly characterized clinically and have been difficult to detect, so there will be likely to be significantly underdiagnosed. Complement deficiencies form about 1-10% of all primary immunodeficiency disorders (Grumach et al 2014). Complement deficiencies can be inherited or acquired. The genetic deficiency of component involved classical pathway (C1q, C1r/s, C2, C4) tend to be linked with autoimmune diseases(Bryan et al 2014), whereas C5 to C9 may have enhanced susceptibility to meningococcal disease.
Complement Deficiencies and Associated Infections
There is a wide variation of infections associated with complement deficiency depending on which complement protein and activation pathway is affected(Skattum et al 2011). Some complement deficiencies carry a risk of infection, others mainly associated with autoimmune diseases. Some genetic and molecular defects have been identified to be associated with complement deficiencies. The deficiencies in the alternative pathway and the terminal pathway is commonly presented with markedly increased susceptibility to Neisserial infections. Properdin deficiency (PD) has been reported in cases of severe meningococcal infections (Fijen et al 1999), recurrent otitis media, and pneumonia (Schejbel et al 2009). PD has been identified as X-linked inheritance. Around 500 published cases of PD with Neisserial infections worldwide. Neisserial meningitidis meningitis and Streptococcal Pneumonie also has been reported incomplete Factor D deficiency. The TP components deficiencies have been reported in association with Neisserial infection. Deficiency of C5, C6, C7 and C8 has less than 1000 reported cases of Neisserial meningitis and sepsis, while C9 deficiencies are more common in Japanese populations with prevalence around 1:1000 (Fukumori et al 1989)(Nagata et al 1989). In LP, MBL deficiencies have been linked to causing recurrent infections in children(super et al, 1989). Around 10- 15% of Caucasians carry the MBL gene that shows a deficient concentration of functional MBL. Another rare deficiency MASP- deficiency, which reported in an adult with autoimmune symptoms and recurrent respiratory infections (Stengaard-Pedersen 2003) and Ficolin-3 deficiency has been described in patients with a recurrent respiratory infection and cerebral abscess (Munthe-Fog et al). Increased susceptibility to encapsulated bacteria, S. pneumonia, and N. meningitidis a common feature of CP deficiencies. CP deficiency also associated with autoimmune disorders, like systemic lupus erythematosus. Few identified genetically determined deficiencies are C2D, C1q, and C4 (Truedsson et al and Sjoholm et al). C2D associated with abnormalities in serum immunoglobulin levels and associated with invasive encapsulated bacteria infection and autoimmune disease (Jonsson et al). Other uncommon deficiencies such as factor H deficiency, the factor I deficiency which associated with susceptibility to encapsulated bacteria along with autoimmune diseases such as glomerulonephritis and vasculitis (Nilsson et al, Nita et al). C1 INH deficiency with a prevalence of approximately 1 in 50 000 presents as hereditary angioedema. Complement deficiencies and associated genetic disorders and clinical manifestations are summarized in Table 1.
Table 1 - Complement deficiencies and clinical associations.
| Proteins |
Genetics |
Frequency |
Infections |
Other associations |
| C1q |
Autosomal recessive |
50-100 |
Sepsis, meningitis, pneumonia,
Streptococcus (S) pneumonia, Neisseria meningitidis |
SLE |
| Cr1/C1s |
Autosomal recessive |
<20 |
Encapsulated bacteria pneumonia, meningitis |
SLE |
| C4 |
Autosomal recessive |
20 -50 |
Sepsis, meningitis, pneumonia,
Streptococcus (S) pneumonia, Neisseria meningitidis |
SLE |
| C2 |
Autosomal recessive |
1/20 000 |
S.pneumonia, staphylococcus aureus, H. Influenza |
SLE |
| C3 |
Autosomal recessive |
20-50 |
Respiratory tract infection, meningitis |
Immune-complex disease |
| Factor D |
Autosomal recessive |
<50 |
Meningitis, sepsis due to neisseria |
|
| Properdin |
X-linked |
100-500 |
Meningitis, sepsis due to neisseria |
|
| MBL |
Common polymorphism in exon 1 |
5-15% of Caucasian |
Respiratory infection |
cardiovascular |
| MASP-2 |
Autosomal recessive |
<20 |
Respiratory infection |
autoimmune |
| Ficolin 3(H-ficolin) |
Autosomal recessive |
1 |
Respiratory infection
Cerebral abscess, Streptococci. H influenza. Pseudomonas aeroginosa |
thrombocytopenia |
| C5 |
Autosomal recessive |
20-50 |
Meningitis due to neisseria |
|
| C6 |
Autosomal recessive |
50-100 |
Meningitis due to neisseria |
|
| C7 |
Autosomal recessive |
50-100 |
Meningitis due to neisseria |
|
| C8 |
Autosomal recessive |
50-100 |
Meningitis due to neisseria |
|
| C9 |
Autosomal recessive |
1/1000 in Japanese |
Meningitis due to neisseria |
|
| Factor H |
Autosomal recessive |
<50 |
Recurrent pyogenic infections N.meningitidis, H influenzae |
Membrano proliferative glomerulonephritis, HUS |
| Factor I |
Autosomal recessive |
5% Caucasian |
Recurrent pyogenic infections N.meningitidis, H influenzae |
Immune complex related disease |
| C1-INH |
Autosomal dominant |
1/50000 |
|
HAE |
| CD59, CD55 |
Somatic mutations of PIG-A |
1-2/1,000,000 |
Cytopenias |
Thrombosis, hematopoetic cytopenia |
| CR3/CD4 (LAD 1) |
Autosomal recessive |
1/1,000,000 |
S. aureus, Gram-negative bacteria |
|
Test for Complement Deficiencies
For the evaluation of particular pathways of the complement system, functional tests have been developed. The ‘total hemolytic complement’ or CH50 is based on a hemolytic assay in which series of patient serum dilutions are incubated with sheep erythrocytes covered with specific IgM, activating the classical complement pathway (Wen L et al). The reciprocal of the serum dilution which causes the lysis of 50% of the erythrocytes is calculated to quantify the amount of active complement. For example, if 50% of the sheep erythrocyte are lysed at a serum dilution of 1:200, this equal toa CH50 value of 200 units/ml. Since the formation of membrane attack complexes (MACs) is necessary for the lysis of the erythrocytes and requires all components of the classical and common terminal complement pathway (C1 through C9), a deficiency or defect in any of these components would result in a significantly lowered CH50. Several variants of this test are in use example one based on liposomes containing enzymes as a read-out to simplify the quantification of lysis using a standard analyzer (Wen et al). Thus, reference values of CH50 may vary depending on the system that is used.
A similar test, the AH50 is used to assess the alternative complement pathway (properdin, factor B, D, and C3) in conjunction with the common terminal pathway (C5 through C9) (Wen et al). Serum dilutions are incubated with rabbit erythrocytes in a buffer that inhibits the classical or lectin pathway. If the alternative pathway is intact, a properdin-stabilized C3 convertase (C3bBb) can form on the surface of the erythrocytes and allow the assembly of C5 convertase and MACs, resulting in lysis which can be quantified.
Using both CH50 and AH50 can help to map the complement deficiency to a specific pathway. If the classical pathway is affected, the CH50 value will be low and the AH50 value will be normal. The opposite is true for a defect in the alternative pathway and if both CH50 and AH50 are low, the abnormal component is likely to be part of the common terminal pathway. However, CH50 and AH50 can also be low due to errors in sample handling because several complement components are unstable (Lock RJ et al). In case of such a result, the quality of the specimens should be controlled and the test might need to be repeated if in doubt.
To test the lectin pathway, a mannan-based ELISA can be used. MBL from the patient’s serum binds the mannan-coated wells, leading to MASP-activation and cleavage of C4. The fragments C4b and C4d can then be quantified using enzymes-linked antibodies (Wen et al).
In order to eventually identify the deficiency, serum levels of individual complement factors can be measured by immunochemical assays based on immunoprecipitation and nephelometry (measurement of turbidity caused by antigen-antibody complexes). Such tests are readily available for C3 and C4. However, quantification of other complement components might require the help pf specialized laboratories. One major drawback of this method is the dependence on the quality of antibodies and standards used. Moreover, functional defects i.e non- functional complement factors with normal serum levels are not detected. Genetic testing by sequencing exons coding for complement components avoids these problems.
Management
Causative therapies are currently unavailable and often unnecessary as standard treatments of complement deficiencies. Replacement of the respective complement components, either recombinant or plasma products, would have too many disadvantages: most complement factors have a short half-life and would require frequent intravenous administration which would be disproportionally inconvenient for the patients. Moreover, they would be costly, possibly immunogenic and plasma products would come with a theoretical risk of transmitting infectious agents. Even in the case of acute infection, replacement therapies are redundant since the patients usually respond to treatment with antibiotics.
Resistance to infections in complement deficient patients can be increased most efficiently by active immunization. Particularly vaccination against encapsulated bacteria is recommended (Jonsson G et al). These include Haemophilus influenza type b, Streptococcus pneumonia, and the main Neisseria meningitides serotypes A, C, W135, Y, and possibly B with the introduction of the new serotype B vaccine. Subsequently, the antibody titers should be controlled to ensure that they are protective. More severe cases with recurrent or persistent serious infections might require prophylactic antibiotic therapy (Kuruvilla M et al)
Concluding Remarks
The role of complement in the immune system has expanded dramatically. It is a well-characterized and an evolving component of host defense, impairment of which leads to susceptibility to infection. Complement represents a cornerstone of the innate defense against infection and provides a vital first-line barrier by activation of proteolytic cascades which leads to the identification and persecution of the surface identified as foreign and allows complement to contain, control, and finally clear invading microorganisms. On top of these important contributions to innate immunity, complement plays a vital role in shaping adaptive immune responses, functionally integrating it into the ability of the host to combat invasion from a wide range of pathogens. The wide range of clinical manifestations of complement deficiencies reflects the complexity of the complement system. As more individuals are identi?ed with specific complement deficiencies, our understanding of how the complement system functions in innate and adaptive immunity will evolve. In conclusion, complement is a multifaceted and robust effector, which bridges the innate and adaptive immune systems. It is vital to host defense, and the extent of its influence is becoming increasingly appreciated as additional information regarding the far-reaching effects of its activation is uncovered. Further study should produce significant knowledge in our understanding of host defense as an integrated process and the roles complement plays in bridging innate and adaptive immunity.
1. Dunkelberger, J.R, Song, WC, 2010 Complement and its role in innate and adaptive immune responses. Cell. Res 20, 34-50.
2. JE Figueroa, Densen ,Infectious diseases associated with complement deficiencies. Clin Microbiol Rev. 1991 Jul; 4(3): 359–395.
3. Hirsch RL, Griffin DE, Winkelstein JA Host modification of Sindbis virus sialic acid content influences alternative complement pathway activation and virus clearance. J Immunol. 1981 Nov;127(5):1740-3.
4. Saifuddin et al. Interaction of MBL with primary isolates of HIV 1. J.gen Viro,81, 949-955, 2000.
5. Grumach .Are complement deficiency really rare. overview on prevalence, importance and modern approach. Molecular Immunology 2014.
6. Angela R. Bryan & Eveline Y. Wu Complement Deficiencies in Systemic Lupus Erythematosus. Curr Allergy Asthma Rep (2014) 14:448.
7. Skattum et al, 2011.Complement deficiency states and associated infections, Mol Immuno 48, 1643-1655.
8. Fijen CA et al,1999. Properdin Deficiency: molecular basis and disease association. Mol. Immunology 36; 863-867.
9. Schejbel et al 2009 Properdin Deficiency in recurrent otitis media and pneumonia. Clin Immnu 131:456-463.
10. Fukumori et al 1989, A high incidence in C9 deficiency among healthy blood donors in Japan. Immuno 1:85-89.
11. Nagata et al 1989, Inherited deficiency of ninth component of complement: an increased risk of meningococcal meningitis. J ped 114,260-264.
12. Stengaard-Pedersen 2003. Inherited deficiency of MBL associated serine protease 2. N Eng J Med 349, 554-560.
13. Munthe-Fog et al 2009. Immunodeficiency associated with FCN3 mutation and ficolin-3 deficiency. New Engl J.Med 360:2637-2644.
14. Truedsson et al 2007. Complement deficiencies and systemic lupus erythematosus. Autoimmunity 40.560-566.
15. Sjoholm et al 2006. Complement deficiencies and diseases; an update. Mol. Immunol 129, 123-131.
16. Jonsson et al 2007. Rheumatological manifestations, organ damage and autoimmunity in hereditary C2 deficiency. Rheumatology ( Oxford)46,1133-1139.